

System validation in the pharmaceutical industry is a crucial process to ensure that computerized systems are fit for their intended use and meet current regulatory requirements. This is a mandatory requirement of cGMP (current Good Manufacturing Practices) and is essential for managing the quality of manufactured products. Any computerized system that could affect Patient Safety, Product Quality, and Data Integrity must be appropriately validated.
Read further or listen to our AI podcast:
Validation aims to build upon existing industry good practice in an efficient and effective manner. It involves demonstrating through documented evidence that each computerized system consistently produces expected results. Key regulatory requirements and guidelines informing validation in this industry include GxP (Good Practice) regulations, EudraLex Volume 4, Annex 11 on Computerised Systems, the US FDA’s 21 CFR Part 11 on Electronic Records and Electronic Signatures, and the ISPE GAMP Guide: A Risk-Based Approach to Complaint GxP Computerized Systems (GAMP 5). GAMP 5, for instance, provides practical guidance, establishes common language, and promotes a system life cycle approach based on good practice.

A computerized system in this context such as MES or EBR systems from Accevo is a set of software and hardware components that together fulfill certain functionalities. The application software should be validated, while the underlying IT infrastructure should be qualified. If a computerized system replaces a manual operation, it should not decrease product quality, process control, or quality assurance, nor should it increase the overall risk of the process.
The V-Model in System Validation
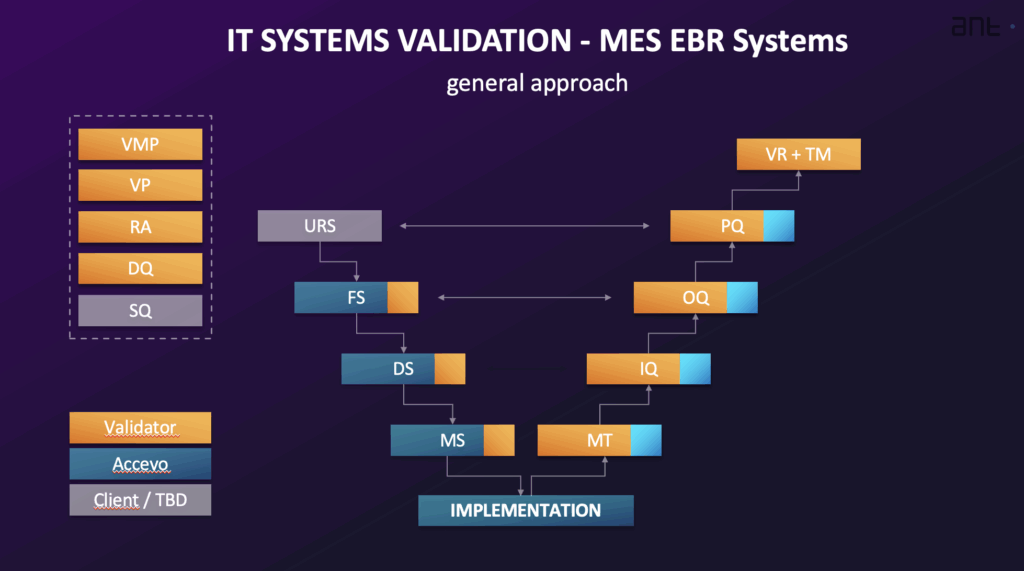
The general approach to validating a computerized system like MES or EBR integrates validation activities with the system’s implementation, typically following a System Life Cycle. This life cycle often includes phases such as Planning and Specification, Design and Development, Verification, Operation, and Retirement. The level of validation effort is commensurate with the system’s complexity (categorization) and the risk associated with its specified intended use (cGxP Criticality). The validation process is often depicted using a V-Model.
Typical validation activities and associated documentation include:
- Validation Master Plan (VMP): A foundational document that provides an overview of the planned validation and qualification activities, defining the scope, framework, quality system standards, roles and responsibilities, approach, and deliverables for validation.
- Validation Plan (VP): Describes the specific activities and deliverables required to implement and operate the system, providing detailed planning, defining responsibilities, and listing necessary documentation. It provides evidence that validation activities will be systematic and controlled.
- User Requirements Specification (URS): Documents the business needs and requirements for system-controlled processes. It specifies what the system must perform. The URS includes general requirements, regulatory requirements (like those for data integrity, audit trail, and security), and functional/process requirements. It should include a list of locations where electronic records and signatures are used and have regulatory impact. Requirements are uniquely identifiable.
- Risk Analysis (RA): A systematic process, often using methods like Simplified FMEA (Failure Mode and Effects Analysis) based on GAMP 5 recommendations, for assessing, controlling, communicating, and reviewing risk throughout the system life cycle. It evaluates the system’s GxP relevance, data integrity, and impact on quality processes, including defining corrective actions. Validation activities are often based on the conclusions of the Risk Analysis.
- Functional Specification (FS) and Software/Hardware Design Specifications (DS): The FS describes the system functions and modules used to meet the requirements defined in the URS. The DS documents how a system is built, including structure, algorithms, logic, data formats, and interface descriptions. The DS also includes configuration and HW/SW design specifications. The FS is created from the URS and any proposal documents and forms the basis for functionality tests. Design Qualification (DQ) involves assessing project documentation to evaluate if the system is designed according to GMP/GAMP rules.
- Supplier Qualification (SQ): Assesses whether the supplier can provide a high-quality product or service, meet regulatory requirements, have adequate quality procedures, and manage implementation, support, and updates.
- Installation Qualification (IQ): Verifies the documented installation and configuration of all system components, both hardware and software, according to specifications. It includes verifying correct installation and configuration.
- Operational Qualification (OQ): Confirms that the system functions as described in the Functional Specification. OQ tests the most critical system functions, especially those managing critical data, and includes positive and negative test results. It should include testing regulatory requirements for electronic records and signatures. OQ is typically performed in a validation environment.
- Performance Qualification (PQ): Verifies and documents that the user requirements are met. PQ tests the overall process managed by the system as defined in the URS. PQ tests are often performed in the production environment using real data.
- Traceability Matrix (TM): Maps all technical requirements from the URS to the corresponding sections of design documents and test procedures (IQ/OQ/PQ). Its goal is to verify that all requirements defined in the URS are met by the system. It demonstrates the relationship between user requirements and successfully executed tests.
- Validation Summary Report (VSR): A document issued at the end of the validation process summarizing all planned activities and deliverables. It analyzes collected data, reports test results (including non-conformities), confirms activities were performed as planned, evaluates test results against acceptance criteria, and provides a clear statement that the system is verified and released for operational use. The Traceability Matrix is typically included as an attachment.
Who Validates?
Validation is carried out by various roles, including the Process owner (responsible for company process, reviewing validation documents, change management), System owner (responsible for technical issues, infrastructure, approving stages), Quality unit (responsible for quality issues, reviewing/approving specific deliverables, ensuring compliance), Project manager (overall responsibility for delivering the solution, reviewing deliverables), and the Supplier (provides system implementation, consultant services, testing).
Maintaining Validation Throughout the System Lifecycle
Validation is not just a one-time activity at implementation. Maintaining the validated state during the system’s operational life and through its retirement phase is also crucial. Activities in these phases include handover, support management, performance monitoring, incident management, CAPA, change management, repair, periodic review, backup and restore, business continuity, security management, system administration, data migration, and system retirement/disposal.
A computerized system in this context is a set of software and hardware components that together fulfill certain functionalities. The application software should be validated, while the underlying IT infrastructure should be qualified. If a computerized system replaces a manual operation, it should not decrease product quality, process control, or quality assurance, nor should it increase the overall risk of the process.
The general approach to validating a computerized system integrates validation activities with the system’s implementation, typically following a System Life Cycle. This life cycle often includes phases such as Planning and Specification, Design and Development, Verification, Operation, and Retirement. The level of validation effort is commensurate with the system’s complexity (categorization) and the risk associated with its specified intended use (cGxP Criticality). The validation process is often depicted using a V-Model.
Typical validation activities and associated documentation include:
- Validation Master Plan (VMP): A foundational document that provides an overview of the planned validation and qualification activities, defining the scope, framework, quality system standards, roles and responsibilities, approach, and deliverables for validation.
- Validation Plan (VP): Describes the specific activities and deliverables required to implement and operate the system, providing detailed planning, defining responsibilities, and listing necessary documentation. It provides evidence that validation activities will be systematic and controlled.
- User Requirements Specification (URS): Documents the business needs and requirements for system-controlled processes. It specifies what the system must perform. The URS includes general requirements, regulatory requirements (like those for data integrity, audit trail, and security), and functional/process requirements. It should include a list of locations where electronic records and signatures are used and have regulatory impact. Requirements are uniquely identifiable.
- Risk Analysis (RA): A systematic process, often using methods like Simplified FMEA (Failure Mode and Effects Analysis) based on GAMP 5 recommendations, for assessing, controlling, communicating, and reviewing risk throughout the system life cycle. It evaluates the system’s GxP relevance, data integrity, and impact on quality processes, including defining corrective actions. Validation activities are often based on the conclusions of the Risk Analysis.
- Functional Specification (FS) and Software/Hardware Design Specifications (DS): The FS describes the system functions and modules used to meet the requirements defined in the URS. The DS documents how a system is built, including structure, algorithms, logic, data formats, and interface descriptions. The DS also includes configuration and HW/SW design specifications. The FS is created from the URS and any proposal documents and forms the basis for functionality tests. Design Qualification (DQ) involves assessing project documentation to evaluate if the system is designed according to GMP/GAMP rules.
- Supplier Qualification (SQ): Assesses whether the supplier can provide a high-quality product or service, meet regulatory requirements, have adequate quality procedures, and manage implementation, support, and updates.
- Installation Qualification (IQ): Verifies the documented installation and configuration of all system components, both hardware and software, according to specifications. It includes verifying correct installation and configuration.
- Operational Qualification (OQ): Confirms that the system functions as described in the Functional Specification. OQ tests the most critical system functions, especially those managing critical data, and includes positive and negative test results. It should include testing regulatory requirements for electronic records and signatures. OQ is typically performed in a validation environment.
- Performance Qualification (PQ): Verifies and documents that the user requirements are met. PQ tests the overall process managed by the system as defined in the URS. PQ tests are often performed in the production environment using real data.
- Traceability Matrix (TM): Maps all technical requirements from the URS to the corresponding sections of design documents and test procedures (IQ/OQ/PQ). Its goal is to verify that all requirements defined in the URS are met by the system. It demonstrates the relationship between user requirements and successfully executed tests.
- Validation Summary Report (VSR): A document issued at the end of the validation process summarizing all planned activities and deliverables. It analyzes collected data, reports test results (including non-conformities), confirms activities were performed as planned, evaluates test results against acceptance criteria, and provides a clear statement that the system is verified and released for operational use. The Traceability Matrix is typically included as an attachment.
Najważniejsze informacje
Validation is carried out by various roles, including the Process owner (responsible for company process, reviewing validation documents, change management), System owner (responsible for technical issues, infrastructure, approving stages), Quality unit (responsible for quality issues, reviewing/approving specific deliverables, ensuring compliance), Project manager (overall responsibility for delivering the solution, reviewing deliverables), and the Supplier (provides system implementation, consultant services, testing).
Validation is not just a one-time activity at implementation. Maintaining the validated state during the system’s operational life and through its retirement phase is also crucial. Activities in these phases include handover, support management, performance monitoring, incident management, CAPA, change management, repair, periodic review, backup and restore, business continuity, security management, system administration, data migration, and system retirement/disposal.
Worth to know! There is a possibility to implement computerized system such as OEE in pharmaceutical industry without validation to support local non-critical system, read more here: Jak wdrożyć system monitorowania OEE bez walidacji w 3 miesiące? - Studium przypadku.
Check also:


